|
|
| Zeile 5: |
Zeile 5: |
| | [[Kategorie:Ernährungslehre]] | | [[Kategorie:Ernährungslehre]] |
| | [[Kategorie:Chemie]][[Kategorie:Chemikalien]] | | [[Kategorie:Chemie]][[Kategorie:Chemikalien]] |
| | + | |
| | + | Gliederung |
| | + | |
| | + | #Proteine |
| | + | #Aminosäuren |
| | + | #Peptide |
| | + | #Entstehung von Peptidbindungen |
| | | | |
| | Proteine | | Proteine |
| Zeile 29: |
Zeile 36: |
| | | | |
| | | | |
| − | Abb. 1: Verknüpfung der AS in Proteinen durch Peptid-Bindung.
| |
| − | Am Aufbau des fortlaufenden Teils der Peptid-Kette (Rückgrat, E backbone) ist also jeder AS-Baustein mit dem gleichen Anteil –CO–C(R)H–NH– beteiligt; nur die außerhalb der Kette liegenden Reste R (Seitenreste, Seitenketten, E side chains) variieren. Mit Hilfe von in den Seitenketten enthaltenen Amino- u. Carboxy-Gruppen bilden einige P. jedoch auch Isopeptid-Bindungen (vgl. Isopeptide) aus.
| |
| − |
| |
| − | Die Polypeptid-Ketten eines makromol. Proteins sind sowohl in Lsg. als auch im Krist. in charakterist. Weise gewunden u. gefaltet u. besitzen unter gegebenen Bedingungen eine ganz bestimmte Konformation. Bei der Struktur der P. unterscheidet man nach Linderstrøm-Lang zwischen Primär-, Sekundär-, Tertiär- u. Quartärstruktur. Für diesen Strukturaufbau sind nicht nur die Säureamid-Bindungen, sondern darüber hinaus kovalente Disulfid-Brücken u. verschiedene Arten von Nebenvalenzbindungen (zwischenmolekulare Kräfte) maßgebend, unter diesen bes. die Wasserstoff-Brückenbindung (in Abb. 2 durch Punktlinien dargestellt).
| |
| − |
| |
| − | Die Primärstruktur wird durch das Zusammentreten der AS zum P. unter Knüpfung der Peptid-Bindung ausgebildet u. ist durch die jeweilige Reihenfolge (Sequenz) der AS charakterisiert. Sie wird durch Sequenzanalyse (s. unten) festgestellt u. beginnend mit der AS, die eine freie a-Amino-Gruppe besitzt (Amino-Terminus, s. Abb. 1) unter Benutzung des Drei- od. Einbuchstabencodes (s. Aminosäuren) angegeben. Man kennt heute über 70 000 P.-Sequenzen. P. mit teilw. übereinstimmenden Primärstrukturen sind meist homolog (s. Homologie); die Übereinstimmung kann aber auch Ausdruck einer funktionell bedingten konvergenten Entwicklung sein.
| |
| − |
| |
| − | Wasserstoff-Brückenbindungen des Typs N–H···O=C zwischen den Atomen des Peptid-Rückgrats sind für die Ausbildung der Sekundärstruktur verantwortlich. Darunter versteht man gewisse regelmäßige, d. h. vom „Zufallsknäuel“ (E random coil) abweichende lokale Faltungsmuster, v. a. die schraubenförmige, rechtsgewundene a-Helix u. das durch parallele od. antiparallele (gegenläufige) Anordnung mehrerer Abschnitte der Polypeptid-Kette zustande kommende b-Faltblatt (Abb. 2).
| |
| − |
| |
| − | Die von Pauling u. Corey aufgeklärte Struktur der a-Helix ist wie folgt zu charakterisieren: 3,6 Aminosäure-Reste pro Windung, 0,54 nm Ganghöhe, 1,05 nm Gesamtdurchmesser. Die Seitenketten weisen nach außen, Wasserstoff-Brücken bilden sich ungefähr in Richtung der Helix-Längsachse. Durch Prolin-Reste wird die Konformation der a-Helix gestört; die Tendenz der einzelnen AS, die a-Helix zu bilden, ist unterschiedlich. Beim b-Faltblatt liegen die Peptid-Ketten in nahezu gestreckter Konformation vor, Seitenreste stehen abwechselnd nach beiden Seiten senkrecht von der Faltblatt-Ebene ab, die Wasserstoff-Bindungen liegen in dieser. Einen hohen Anteil an a-Helix besitzen z. B. a-Keratine, Myosine, Hämoglobin, Myoglobin, während Fibroin, Immunglobuline u. a. überwiegend aus b-Faltblatt bestehen. Eine zylindr. Anordnung von 8 parallelen b-Faltblatt-Strängen, die von 8 a-Helices umgeben sind, verleiht dem sog. a/b-Faß (E a/b barrel) der Triosephosphat-Isomerase, der Ribulosebisphosphat-Carboxylase u. vieler anderer P. seinen ästhet. Reiz; ein reines b-Faß (10 zylindr. angeordnete b-Stränge, abwechselnd „auf- u. abwärts“ laufend) liegt z. B. bei Superoxid-Dismutase vor, ein entsprechendes 8-strängiges b-Faß ziert die Lipocaline. Gewisse Sklero-P. besitzen bes. Sekundärstrukturen wie z. B. 3 umeinander gewundene linksgängige Helices bei Collagenen; dimerisierende P. haben zuweilen zwei miteinander verdrillte a-Helices (E: coiled coil) . Eine nicht selten vorgefundene Ungleichverteilung von polaren u. unpolaren AS auf zwei Längshälften der a-Helix resultiert in der amphipathischen Helix.
| |
| − |
| |
| − | Unter Tertiärstruktur versteht man die räumliche Anordnung der Peptid-Kette sowie der AS-Seitenreste, die durch Disulfid-Brücken, durch Wasserstoff-Brückenbindungen, durch ion. u. durch hydrophobe Wechselwirkungen (s. hydrophobe Bindung), meist zwischen AS-Seitenketten, stabilisiert wird. Durch Disulfid-Brücken können – wie bei Ribonuclease u. Chymotrypsinogen – in der Sequenz voneinander entfernte Bereiche einer Polypeptid-Kette od. – wie bei Chymotrypsin u. Immunglobulin – mehrere Polypeptid-Ketten kovalent miteinander verbunden werden. Im Insulin finden sich zwei Disulfid-Bindungen zwischen den beiden Polypeptid-Ketten u. eine dritte zwischen den AS-Resten 6 u. 11 der sog. A-Kette. Die Wasserstoff-Brückenbindung zwischen AS-Seitenketten, die in Ggw. von Wasser, d. h. an der Oberfläche des Makromol., eher instabil ist, besitzt ihre größte Bedeutung in dessen Inneren. Dort befinden sich aufgrund hydrophober Wechselwirkung v. a. unpolare (hydrophobe, lipophile) AS-Reste, während sich die polaren u. geladenen Seitenketten (letztere können ion. Bindungen eingehen) in wäss. Lsg. eher nach außen wenden. Im Fall der integralen Membran-P. (s. Membranen) besteht jedoch der Teil der P.-Oberfläche, der ins lipophile Milieu der Membran eingebettet ist, ebenfalls überwiegend aus unpolaren AS-Gruppen. Auch ohne Disulfid-Brücken können allein durch Nebenvalenz-Stabilisierung mehrere Polypeptid-Ketten zu einer funktionellen Einheit verbunden sein. Eine Quartärstruktur liegt dann vor, wenn ein P. nicht aus einer einzigen Polypeptid-Kette besteht, sondern aus einer definierten Anzahl solcher Ketten (Untereinheiten; häufig 4, z. B. bei Hämoglobin, aber auch 2, 6 od. 20), die untereinander nicht durch Peptid-Bindungen, sondern durch intermol. wirkende Kräfte zusammengehalten werden. Dabei können sich Konformationsänderungen der einen Untereinheit den übrigen mitteilen u. auch bei diesen zu Veränderungen führen (Kooperativität, Beisp.: Hämoglobin).
| |
| − |
| |
| − | Die vier genannten Strukturniveaus (Primär-, Sekundär-, Tertiär- u. Quartärstruktur) sind voneinander abhängig. So kann man – wenn auch noch mit mangelnder Treffsicherheit – die Elemente der Sekundärstruktur (Helix, Faltblatt) eines bestimmten P. aus dessen AS-Sequenz ableiten. Im Prinzip sollte es sogar möglich sein, die Gesamtstruktur aus der Kenntnis der Primärstruktur vorherzusagen, jedoch ist dieser Anspruch bis heute noch nicht verwirklicht . Einige nicht natürlich vorkommende P. konnten jedoch am Reißbrett entworfen u. mit den gewünschten Struktur-Eigenschaften synthetisiert werden. Durch Kristallstrukturanalyse u. mehrdimensionale NMR-Spektroskopie (vgl. im Abschnitt über Analytik) kennt man heute jedoch die Geometrien hunderter verschiedener P.-Typen. Die vorliegenden Daten (zu einer P.-Datenbank s. Protein Data Bank) gestatten nicht nur, durch Strukturvergleich den Ablauf der biolog. Evolution nachzuvollziehen, sondern auch den Mechanismus der P.-Faltung , die spontan erfolgen od. durch Chaperone assistiert werden kann, zu erforschen – schließlich möchte man verstehen, wodurch die Topologie eines P. bestimmt wird.
| |
| − |
| |
| − | Es hat sich erwiesen, daß sich bestimmte Teilbereiche der Kette mehr od. weniger unabhängig voneinander falten, wodurch sich Domänen bilden. Die Raumstruktur der P. darf man sich übrigens nicht vollkommen starr vorstellen. Ihre Flexibilität, die sich in Fluktuationen zwischen Konformations-Unterzuständen u. mol. Beben ausdrückt u. die auch z. B. durch NMR-Spektroskopie festgestellt werden kann, ermöglicht es erst den Makromol., ihre Funktionen als Enzyme, Rezeptoren usw. zu erfüllen, u. ist auch unerläßlich für die Übertragung von Konformationsänderungen, die alloster. Regulation (s. Allosterie) u. die Wechselwirkungen von P. untereinander (z. B. Protease u. Inhibitor) od. mit anderen Makromol. wie Nucleinsäuren (z. B. Transkriptionsfaktor u. Desoxyribonucleinsäure, Abk.: DNA). Zur Veranschaulichung solcher mol. Wechselwirkungen u. Dynamik sowie zu Energieberechnungen bemüht man in der biochem., molekularbiolog. u. pharmakolog. Forschung Computer-Simulationen (E molecular modeling bzw. molecular dynamics simulation).
| |
| − |
| |
| − | Biosynth.: Die Biosynth. der P. aus AS od. Translation findet in eukaryo(n)tischen Zellen an den Ribosomen (vgl. dort) des Cytoplasmas, des rauhen endoplasmatischen Retikulums (ER) u. der Kernhülle statt. Etwas andersgeartete Ribosomen besitzen die Bakterien, die Mitochondrien u. Plastiden. Die genannten Zell-Organellen besitzen also auch die Fähigkeit zur P.-Synth., importieren daneben aber viele P. aus dem Cytoplasma.
| |
| − |
| |
| − | Zu den einzelnen Schritten der P.-Biosynth. s. Translation.
| |
| − | Die Synth. der P. beginnt am Amino-Ende u. endet am Carboxy-Terminus. Man nimmt an, daß sie sich schon während des Synth.-Vorgangs zu falten beginnen. Viele sekretor. P. tauchen – auch schon während ihrer Synth. – mit ihren aminoterminalen Erkennungssequenzen (Signalpeptiden) in die Membran des ER ein u. werden dort eingeschleust . Dabei spielt ein bestimmtes Nucleoprotein, die Signal-Erkennungs-Partikel (E signal recognition particle, Abk. SRP), eine Vermittler-Rolle. P., die anschließend im ER zu verbleiben haben, besitzen das Retentions-Signal KDEL (Lys-Asp-Glu-Leu). Im Cytoplasma synthetisierte, für den Import z. B. in Mitochondrien vorgesehene P. besitzen ebenfalls Signal-Sequenzen u. werden durch die äußere u. die innere Membran, u. zwar an deren Kontaktstellen transportiert. Haben diese P. die Membranen passiert, werden sie durch spezif. Proteasen ihres Signalpeptids entledigt, wodurch der Transport irreversibel wird, u. falten sich mit Unterstützung durch das Hitzeschock-Protein hsp60. Für die innere Mitochondrien-Membran od. den Membran-Zwischenraum bestimmte P. besitzen weitere Signalsequenzen. Noch komplizierter ist der P.-Transport in Chloroplasten , da bei diesen zusätzlich das Kompartiment der Thylakoiden als Bestimmungsort in Frage kommt. In den Zellkern gelangen P., die die dementsprechende Signalsequenz besitzen, durch die Kernporen mit Hilfe des Kernporen-Komplexes u. verschiedener lösl. Proteine wie Importin u. des kleinen GTP-bindenden Proteins Ran.
| |
| − |
| |
| − | Während der Translation (cotranslational) finden mit der Acetylierung u./od. Entfernung eines Methionin-Rests bei Eukaryonten bereits Modifizierungen des Amino-Terminus des entstehenden (naszierenden) P. statt. Im ER u. im Golgi-Apparat, über den die P. per Vesikel-Transport zum Bestimmungsort wandern (z. B. Cytoplasma-Membran, Vakuole, Lysosomen), aber auch im Cytoplasma erfolgen etliche posttranslationale Modifikationen, darunter Acylierung, Carboxylierung (in Pos. 4 von Glutaminsäure-Resten – Vitamin-K-abhängig), Glykosylierung, Glypiierung (Anknüpfung eines Glykosylphosphatidylinosit-Ankers), Prenylierung (s. Prenylproteine), Disulfid-Isomerisierung (durch P.-Disulfid-Isomerase, EC 5.3.4.1), Hydroxylierung (von Lysin u. Prolin – Vitamin-C-abhängig, s. Collagene), Phosphorylierung (an Threonin, Serin, Tyrosin; s. a. Protein-Kinasen), Sulfatierung (an Tyrosin), Protein-Spleißen (vgl. Inteine) u. andere. Proteohormone u. sekretor. Enzyme müssen schließlich noch aus (nicht od. anders aktiven) Vorstufen (Prohormonen bzw. Proenzymen od. Zymogenen) „herausgeschnitten“ werden.
| |
| − |
| |
| − | Abbau: Von außen mit der Nahrung zugeführte P. werden im Verdauungstrakt, körpereigene dagegen meist intrazellulär zu AS abgebaut. Bei der Verdauung erfolgt im Magen u. Darm eine Aufspaltung in Peptide bzw. AS unter dem Einfluß Eiweiß-spaltender Enzyme (Proteasen), die allerdings zuvor erst aus ihren Zymogenen freigesetzt werden müssen. Die Spaltprodukte wandern durch die Darmwand u. werden in den arbeitenden Zellen (nach erfolgter Desaminierung) zu Kohlendioxid u. Wasser oxidiert (Katabolismus) od. aber mit Hilfe von Nucleinsäuren u. Enzymen zu arteigenen Eiweißstoffen zusammengefügt (Anabolismus). Beim vollständigen Abbau ergibt 1 g P. die Energie von etwa 17,2 kJ (4,1 kcal).
| |
| − |
| |
| − | Durch oxidative Prozesse u. Glykation altern die Proteine. Die menschlichen Eiweißstoffe der Leber werden in 10–20 Tagen, diejenigen der Haut u. Muskulatur in ca. 160 Tagen zur Hälfte erneuert. Die Hälfte des menschlichen Bluteiweißes wird in 10 Tagen ab- u. wieder aufgebaut, u. täglich werden 9% der Plasma-Albumine umgesetzt. Bei Eukaryonten werden intrazelluläre P., die im Cytoplasma abgebaut werden sollen, von einem Multienzym-Komplex an ihrem aminoterminalen AS-Rest sowie einer AS im Inneren der Kette erkannt u. mit mehreren Mol. des Polypeptids Ubiquitin verknüpft. Dies ist das „Brandzeichen“ für einen Adenosin-5'-triphosphat-abhängigen Protease-Komplex (Proteasom), der die ubiquitinierten P. verdaut. Bei Proteolyse innerhalb von tier. Zellen spielen auch Lysosomen u. die darin enthaltenen Kathepsine eine wichtige Rolle. Dabei werden P., die bestimmte AS-Sequenzen enthalten, schneller abgebaut als andere.
| |
| − |
| |
| − | Ernährung: Bei der Nutzung der P. denkt man zunächst an die Ernährung von Mensch u. Tier. Unter allen Nahrungsmitteln kann das P. dabei am wenigsten entbehrt werden. Durch gesteigerte Eiweiß-Verbrennung läßt sich ein Ausfall an Fetten u. Kohlenhydraten für einige Zeit ausgleichen, dagegen erfolgt bei völlig fehlender P.-Zufuhr (selbst bei überreichlicher Zufuhr an Fett u. Kohlenhydraten) eine nach kurzer od. längerer Zeit tödliche Auszehrung, da der Erwachsene täglich ca. 30 g seines Körpereiweißes verbrennt. Der tägliche Mindestbedarf an P. (Milcheiweiß) wird von der WHO auf 37 g für einen Mann von 65 kg u. auf 29 g für eine Frau von 55 kg berechnet; es gibt allerdings auch Auffassungen, daß diese Werte zu niedrig angesetzt seien. Man erwartet im allg., daß etwa 15% des Brennwertbedarfs durch P. gedeckt werden.
| |
| − |
| |
| − | Übrigens sind die P. der verschiedenen Nahrungsmittel wegen unterschiedlicher AS-Zusammensetzung für den Menschen biolog. nicht gleichwertig. Wenn man z. B. für Milcheiweiß die Vergleichszahl 100 setzt, ergibt sich für P. aus Rindfleisch 104, für Fisch-P. 95, Reis-P. 88, Kartoffel-P. 79, Erbsen-P. 55 u. für Weizenmehl-P. 40. Der biolog. Wert von Hefe-P. liegt zwischen dem von tier. u. pflanzlichem Eiweiß. Der Mensch kann also z. B. aus 100 g Fleisch-P. bedeutend mehr körpereigene Substanz aufbauen als etwa aus 100 g Weizen-P. od. Mais-Protein. Mangel an P. führt bes. bei Kindern der P.-armen, feuchten Tropengebiete oft zu ausgesprochenen, manchmal tödlichen P.-Energie-Mangelsyndromen (PEM) wie Marasmus (kalor. Unterernährung) od. Kwashiorkor (Mehlnährschaden). Kwashiorkor wird heute zusätzlich auf eine Schädigung der Leber durch Aflatoxine als Ursache zurückgeführt.
| |
| − |
| |
| − | Herst.: Chem. Synth.: Die laboratoriumsmäßige Teilsynth. von P. ist schon Emil Fischer vor dem 1. Weltkrieg geglückt. Er konnte bereits einen Eiweiß-ähnlichen Körper aus 18 Aminosäuren mit einer MR von rund 1200 aufbauen (Peptid-Synthese). Zahn et al. gelang 1963 die erste Totalsynth. eines P. (Insulin), u. 1969 synthetisierten Gutte u. Merrifield (Nobelpreis für Chemie 1984) in 11 931 Schritten in ihrer Synth.-Maschine (s. Merrifield-Technik) die gesamte Sequenz der Ribonuclease. Zur Synth. von Peptiden im Labormaßstab ist die Meth. durchaus eingeführt, für P. ist sie jedoch im allg. immer noch relativ aufwendig u. besitzt vergleichsweise geringe Bedeutung. Natürlich kommen derartige vollsynthet. Meth. für die Gewinnung von Nahrungs-P. erst recht nicht in Frage.
| |
| − |
| |
| − | In zunehmendem Maß werden heute die Meth. der Gentechnologie (Näheres s. dort) zur Synth. bestimmter P. angewendet (E protein engineering; z. B. Interferon, Somatostatin, Insulin), bei denen man sich der das betreffende P. codierenden DNA bedient, indem man diese entweder durch Total- od. Teilsynth. bereitstellt bzw. cDNA verwendet od. natürlich vorkommende DNA modifiziert (gezielte Mutagenese, E site directed mutagenesis).
| |
| − |
| |
| − | Angesichts der großen biolog. u. ernährungsphysiolog. Wichtigkeit der Eiweißstoffe kommt der ausreichenden P.-Produktion bes. Bedeutung zu. Schon seit Jahren hält man daher nach möglichen Quellen für die zukünftige P.-Versorgung durch unkonventionelle Meth. Ausschau. Grundsätzlich bieten sich hier zwei Möglichkeiten an: 1. Chem./biochem./biotechnolog. u. 2. biolog. Methoden.
| |
| − | Bei den biochem. Verf. ließ man zunächst Zucker (Glucose, Xylose) u. Stickstoff-haltige Nährsalze mit Hilfe bestimmter Wildhefen (z. B. Torula-Hefen) statt zu Alkohol zu P. „vergären“. Später fand man durch systemat. Untersuchungen – Arbeitsgebiet ist die Biotechnologie – weitere Substrate, die sich zusammen mit Stickstoff-, Schwefel- u. Phosphor-Quellen zur P.-Gewinnung eigneten, z. B. Cellulose, Sulfit-Ablaugen, Melasse u. a. Abfallprodukte, Erdöl-Paraffine (zur Deckung des Weltbedarfs an P. aus Erdöl würden weniger als 2% der Welt-Erdölförderung genügen), Ethanol, Methan u. Methanol. Als zur mikrobiellen P.-Herst. geeignete Organismen erwiesen sich neben Hefen auch Algen u. Bakterien, also einzellige Mikroorganismen. Daher bezeichnet man die so produzierten P. heute bevorzugt als Einzellerproteine (EZP, s. single cell protein, Abk. SCP). Zwar haben sich die ursprünglichen Hoffnungen hinsichtlich der großtechn. Gewinnung von Nahrungs-P. bisher nicht erfüllt – die meisten P.-Qualitäten sind wegen ihres Aminosäure-Ungleichgew. u. erhöhten Nucleinsäure-Gehalts nicht ohne weiteres für den menschlichen Verzehr geeignet –, doch dürfte das SCP auf dem Umweg über Futtermittel beim Ausgleich des globalen P.-Defizits von Nutzen sein. Einschränkend ist festzustellen, daß z. Z. die Herst. von P. aus Erdöl (Petroproteine), verglichen mit derjenigen aus Methan(ol) od. gar aus Soja- od. Fischmehl (E fish protein concentrate, FPC), nicht wirtschaftlich ist.
| |
| − |
| |
| − | Im Gegensatz zu den SCP sind P. aus pflanzlichen Quellen, ggf. nach Supplementierung defizitärer Aminosäuren (Fortifikation) u. nach Texturierung (vgl. Textur), direkt für die menschliche Ernährung geeignet. Aus Sojabohnen lassen sich die als textured vegetable proteins (TVP) bekanntgewordenen, fleischähnlichen P.-Produkte gewinnen, u. ähnlich ist das von Courtaulds entwickelte edible spun protein (KESP), das aus Puff- od. Saubohnen hergestellt wird. Als weitere pflanzliche P.-Quellen kommen in Frage: Baumwollsamen, Sonnenblumen, Sesam, Raps, Leinsamen, Luzerne, Lupinen, Erdnüsse u. selbst Gräser (leaf protein concentrate, LPC).
| |
| − |
| |
| − | Die sog. biolog. Meth. der P.-Synth. bedienen sich der systemat. u. kontrollierten Züchtung von Fischen, Krebsen (z. B. Krill), Geflügel u. fleischliefernden Säugetieren bzw. der qual. u. quant. Verbesserung P.-liefernder Pflanzen durch züchter. Maßnahmen.
| |
| − | Reinigung: Die Isolierung bestimmter P. aus biolog. Material u. ihre Reinigung erfolgt klass. durch fraktionierte Fällung mit Salzen (z. B. Ammoniumsulfat) od. organ. Lsm. (z. B. Aceton), durch Adsorption (z. B. an Hydroxylapatit), durch Ionenaustausch- u. Gelchromatographie, verschiedene Elektrophorese-Verf., präparative Ultrazentrifugation. Sie wird erleichtert durch neue Entwicklungen bei der HPLC (fast protein liquid chromatography, FPLC), od. durch Chromatofokussierung (eine Kombination der Säulenchromatographie u. der isoelektrischen Fokussierung). Ein sehr effizientes Mittel ist oft die Affinitätschromatographie.
| |
| − |
| |
| − | Analytik : Zum qual. u. teilw. quant. Nachw. sind folgende Reaktionen geeignet: Biuret-, Kjeldahlsche, Lowrysche, Millonsche, Ninhydrin-, Paulysche u. Xanthoprotein-Reaktion. Die UV-Photometrie macht Gebrauch von der Lichtabsorption durch aromat. AS-Reste.
| |
| − | Ermittlung der Mol.-Größe u. Form: Die Molmassen der makromol. P. können in Ultrazentrifugen bestimmt werden, od. sie lassen sich aus Lichtstreuung berechnen: P.-Lsg. sind opaleszierend (Tyndall-Effekt), u. aus der Intensität des gestreuten Lichts läßt sich auf die Molmasse schließen. Als Schnellmeth. hat sich die Gelelektrophorese bewährt; daneben findet die Gelchromatographie (Gelpermeationschromatographie, Gelfiltration) Anwendung. Bei P.-Krist. läßt sich das Mol.-Gew. recht genau durch Röntgenmessungen ermitteln, u. wertvolle Aufschlüsse liefert auch die Massenspektrometrie.
| |
| − |
| |
| − | Messungen der Viskosität u. Strömungsdoppelbrechung haben ergeben, daß Hämoglobin, Globulin, Ovalbumin kugelförmige od. rotationsellipsoide Mol. von Kolloidgröße, Fibrinogen, Myosin, Kollagen u. Seidenfibroin langgestreckte, Wollkeratin dagegen zickzackartig gefaltete Mol. besitzen. Mit dem Elektronenmikroskop kann man zahlreiche P.-Komplexe sichtbar machen; z. B. Tabakmosaikvirus, ATP-Synthase, Pyruvat-Dehydrogenase-Komplex. Zur Untersuchung von P. durch Infrarot-Spektroskopie s. Lit. .
| |
| − |
| |
| − | Aminosäure-Analyse: Zur quant. Bestimmung der Zusammensetzung eines P. aus AS müssen diese zunächst hydrolyt. freigesetzt werden. Am häufigsten wird zu dieser Proteolyse Säure verwendet. Man erhitzt dazu das P. 20 h od. länger mit der mehrfachen Menge seines Gew. an 6 M Salzsäure im geschlossenen Röhrchen auf 110 °C. Die heute meist automatisierte Trennung des AS-Gemisches erfolgt nach Moore u. Stein (Nobelpreis für Chemie 1972) durch Ionenaustauschchromatographie auf sulfonyliertem Polystyrol bei steigenden pH-Werten (Moore-Stein-Analyse). Automat. Nachw. der AS durch Ninhydrin.
| |
| − |
| |
| − | Sequenzanalyse : Nach der AS-Analyse u. der Endgruppenbestimmung (s. dort) werden ggf. die verschiedenen Polypeptid-Ketten unter reduktivem Aufbrechen der Disulfid-Brücken getrennt. Einzeln werden sie spezif. in Teil-Peptide „zerschnitten“, z. B. enzymat. mit Trypsin, das an der Carboxy-Gruppe von Lysin u. Arginin angreift, od. chem. mit Bromcyan an Methionin. Das Ziel ist, ausreichend kurze Peptide zu erhalten, deren Sequenzen sich in bezug auf die Gesamtsequenz überlappen, so daß eindeutig auf letztere geschlossen werden kann. Zur Ermittlung der AS-Sequenzen sind spezif. schrittweise arbeitende u. daher mechanisierbare bzw. automatisierbare Abbaumeth. entwickelt worden (v. a. der Edman-Abbau, s. a. Sequenzanalyse), die den Zeitaufwand für die Strukturermittlung von P. sehr stark zu reduzieren vermögen.
| |
| − |
| |
| − | Ermittlung der Raumstruktur: Der Helix-Anteil der Sekundärstruktur kann aus Circulardichroismus u. opt. Rotationsdispersion abgeschätzt werden. Bei der Klärung der räumlichen Struktur von P. hat die Kristallstrukturanalyse mit Röntgen- u. Neutronen-Beugung bes. wichtige Aufschlüsse geliefert (erreichte Auflösung: ca. 0,2 nm). Kendrew u. Perutz (Nobelpreis für Chemie 1962) lieferten die ersten Röntgenstrukturen von P.
| |
| − | Peptid-Bindung
| |
| − |
| |
| − | Carbonsäureamid-Gruppierung –CO–NH–, die bei der Kondensation von Aminocarbonsäuren entsteht, wenn die Carboxy-Gruppe (–COOH) einer Aminosäure mit der Amino-Gruppe (–NH2) einer anderen Aminosäure unter Wasserabspaltung reagiert. Die P.-B. ist das strukturelle Charakteristikum der Peptide (Oligo-, Polypeptide) u. Proteine. Wie andere Amid-Bindungen auch, unterliegt die P.-B. folgender Mesomerie:
| |
| − |
| |
| − |
| |
| − |
| |
| − | Daraus resultiert zum einen ihre relative chem. Stabilität u. zum andern eine eingeschränkte Drehbarkeit um die C–N-Achse infolge eines partiellen Doppelbindungscharakters; die 4 an der P.-B. beteiligten Atome sowie die direkt daran gebundenen a-Kohlenstoff-Atome liegen in einer Ebene – meist in trans-Konfiguration, außer Amino-seitig von L-Prolin, wo unter Mithilfe der Peptidylprolyl-cis-trans-Isomerase (PPIase, EC 5.2.1.8) cis-P. B. gebildet werden können. Die Beschränkungen der Drehbarkeit sind von Bedeutung für die Sekundärstruktur der Proteine. Die Knüpfung der P.-B. erfolgt bei der Protein-Biosynth. in den Ribosomen, kann aber auch durch andere Enzyme (z. B. normalerweise Peptid-spaltende wie z. B. Papain) katalysiert werden. Sind an P.-B. andere Amino-Gruppen als a-Amino-Gruppen u./od. andere Carboxy-Gruppen als a-Carboxy-Gruppen beteiligt, spricht man von Isopeptid-Bindungen, s. Isopeptide. Diese werden nicht während der ribosomalen Protein-Synth., sondern in anderen Transferase-Reaktionen gebildet. Der Aufbau der P.-B. in vitro wird bei Peptid-Synthese erläutert. Die Spaltung der P.-B. kann chem. (z. B. durch halbkonz. Salzsäure, Hydrazin) od. enzymat. (durch Peptidasen) erfolgen.
| |
| − |
| |
| − |
| |
| − | E peptide linkage
| |
| − | F liaison peptidique
| |
| − | I legame peptidico
| |
| − | S enlace peptídico
| |
| − |
| |
| − |
| |
| − | Quelle: Römpp Lexikon Chemie – Version 2.0, Stuttgart/New York: Georg Thieme Verlag 1999
| |
| | | | |
| | Peptide | | Peptide |
| Zeile 111: |
Zeile 48: |
| | Biolog. Bedeutung: Auf die Bedeutung der makromol. P. für pflanzliche u. tier. Organismen wird bei Proteine ausführlich eingegangen. Eine gleichermaßen spezif. Rolle spielen Oligo- u. Polypeptide im tier. Organismus z. B. als Hormone (Peptidhormone), Wachstumsfaktoren, Cytokine, Neurotransmitter u. Neuromodulatoren (Neuropeptide). Für die physiolog. Wirkung der P. ist neben der Konfiguration die Konformation u. die mol. Dynamik von Bedeutung, u. natürlich benötigen die P., um als Mediatoren wirksam werden zu können, spezif. Rezeptoren. Bei der Zell-vermittelten Immunantwort werden Antigene (Fremd-Proteine) von Antigen-präsentierenden Zellen zu P. (Antigen-Peptide, T-Zell-Epitope) abgebaut, von Histokompatibilitäts-Antigenen komplexiert u. so an der Zelloberfläche den T-Lymphocyten zum „Abtasten“ dargeboten; von außen verabreichte P. (peptide feeding) werden ebenfalls präsentiert. Auch von körpereigenen Proteinen abgeleitete Selbst-P. werden präsentiert, was in der Frühphase der T-Zell-Entwicklung für die Entstehung von Selbst-Toleranz von Bedeutung ist. P.-Ester können für süßen (Aspartame®) od. bitteren Geschmack verantwortlich sein, u. wieder andere P. treten als Toxine pflanzlichen od. tier. Ursprungs in Erscheinung. Auch unter den Antibiotika finden sich eine Reihe von P. (Peptid-Antibiotika ), die z. T. Aminosäuren der „unnatürlichen“ D-Konfiguration enthalten, ggf. auch Hydroxycarbonsäuren, die über Esterbindungen verknüpft sind (Peptolide). Viele der physiolog. aktiven P. liegen als homodete od. heterodete Cyclopeptide vor. | | Biolog. Bedeutung: Auf die Bedeutung der makromol. P. für pflanzliche u. tier. Organismen wird bei Proteine ausführlich eingegangen. Eine gleichermaßen spezif. Rolle spielen Oligo- u. Polypeptide im tier. Organismus z. B. als Hormone (Peptidhormone), Wachstumsfaktoren, Cytokine, Neurotransmitter u. Neuromodulatoren (Neuropeptide). Für die physiolog. Wirkung der P. ist neben der Konfiguration die Konformation u. die mol. Dynamik von Bedeutung, u. natürlich benötigen die P., um als Mediatoren wirksam werden zu können, spezif. Rezeptoren. Bei der Zell-vermittelten Immunantwort werden Antigene (Fremd-Proteine) von Antigen-präsentierenden Zellen zu P. (Antigen-Peptide, T-Zell-Epitope) abgebaut, von Histokompatibilitäts-Antigenen komplexiert u. so an der Zelloberfläche den T-Lymphocyten zum „Abtasten“ dargeboten; von außen verabreichte P. (peptide feeding) werden ebenfalls präsentiert. Auch von körpereigenen Proteinen abgeleitete Selbst-P. werden präsentiert, was in der Frühphase der T-Zell-Entwicklung für die Entstehung von Selbst-Toleranz von Bedeutung ist. P.-Ester können für süßen (Aspartame®) od. bitteren Geschmack verantwortlich sein, u. wieder andere P. treten als Toxine pflanzlichen od. tier. Ursprungs in Erscheinung. Auch unter den Antibiotika finden sich eine Reihe von P. (Peptid-Antibiotika ), die z. T. Aminosäuren der „unnatürlichen“ D-Konfiguration enthalten, ggf. auch Hydroxycarbonsäuren, die über Esterbindungen verknüpft sind (Peptolide). Viele der physiolog. aktiven P. liegen als homodete od. heterodete Cyclopeptide vor. |
| | | | |
| − | Analytik : Zum qual. Nachw. von P. sind einige der auch auf Aminosäuren anwendbaren Meth. geeignet, ferner die Biuret-Reaktion, die zusammen mit Folins Reagenz (s. dort Punkt 4) auch zur quant. Bestimmung geeignet ist (Lowry-Methode). Die Bestimmung der Aminosäure-Zusammensetzung von P. ist erst nach hydrolyt. Spaltung möglich, die chem. od. enzymat. mit Proteasen vorgenommen werden kann. Hochauflösende Auftrennungen u. Charakterisierungen von P.-Gemischen können mit HPLC, Kapillarelektrophorese u. Massenspektrometrie erfolgen. Zur Trennung der Aminosäuren bedient man sich chromatograph. Meth. (Dünnschicht-, Gas- u. Ionenaustauschchromatographie, HPLC). Die Ionenaustauschchromatographie hat bes. breite Anw. gefunden u. ist als Moore-Stein-Analyse automatisiert worden. Einen Aufschluß über den tatsächlichen Aufbau von P., d. h. über die Art der Verknüpfung der Aminosäure-Bausteine miteinander, erhält man aber erst durch die Sequenzanalyse, denn schon 2 Aminosäuren (z. B. Glycin u. Alanin) können ja zu zwei verschiedenen Dipeptiden (s. oben) zusammentreten. Die Sequenzanalyse ist prinzipiell eine Meth. der Endgruppenbestimmung, bei der die Peptid-Kette wiederholt an einem Ende (meist der freien Amino-Gruppe) um jeweils einen Aminosäure-Rest verkürzt wird (z. B. mit Aminopeptidasen). Zum Markieren der Endgruppe führte Sanger 1945 bei der Insulin-Analyse das 1-Fluor-2,4-dinitrobenzol ein, das sich mit den endständigen Aminosäuren zu 2,4-Dinitrophenyl(Dnp)-Aminosäuren umsetzt, die nach Hydrolyse einzeln nachweisbar sind. Eine Weiterentwicklung ist die Dansylchlorid-Meth., bei der Dansyl-Aminosäuren anfallen. Da sich beim P.-Abbau die Reaktionsschritte wiederholen, sind schon früh Ansätze zur Mechanisation u. Automation der Abläufe gemacht worden. Insbes. für den Edman-Abbau sind selbständig arbeitende Geräte in Benutzung, die den Zeit- u. Substanzbedarf für eine P.-Sequenzanalyse auf einen Bruchteil des früher benötigten reduzieren – Sequenzanalysen sind heute schon mit Nanogramm-Mengen (pmol-Bereich) möglich. Bei der Analyse der Primärstruktur der P., wie die Aminosäure-Sequenz auch bezeichnet wird, leistet auch die Massenspektrometrie gute Dienste. Die Untersuchung der Sekundär- bis Quartärstrukturen (Näheres s. Proteine) bedient sich vorwiegend physikal. Meth. wie des Circulardichroismus, der Röntgenstrukturanalyse od. NMR-Spektroskopie.
| |
| | | | |
| | Herst.: Auch bei der Synth. ist der zeitliche Aufwand aufgrund der Entwicklung automat. Verf. u. der Festphasen-Technik (Merrifield-Technik) ungleich geringer geworden. Für die Herst. biolog. aktiver u. pharmakolog. nutzbarer P. werden heute neben der chem. Peptid-Synthese in zunehmendem Maße Meth. der Biotechnologie u. Gentechnologie eingesetzt, was z. B. auf dem Gebiet der Peptidhormone bereits zu Erfolgen geführt hat. | | Herst.: Auch bei der Synth. ist der zeitliche Aufwand aufgrund der Entwicklung automat. Verf. u. der Festphasen-Technik (Merrifield-Technik) ungleich geringer geworden. Für die Herst. biolog. aktiver u. pharmakolog. nutzbarer P. werden heute neben der chem. Peptid-Synthese in zunehmendem Maße Meth. der Biotechnologie u. Gentechnologie eingesetzt, was z. B. auf dem Gebiet der Peptidhormone bereits zu Erfolgen geführt hat. |
| Zeile 117: |
Zeile 53: |
| | Biosynth.: Meist durch enzymat. „Resektion“ aus Proteinen, die nach Maßgabe des genetischen Codes (Näheres s. dort) u. der Sequenzinformation der Messenger-Ribonucleinsäuren in den Ribosomen gebildet werden, vgl. Peptidhormone. In manchen Fällen findet jedoch durch nicht-ribosomale Enzyme eine Biosynth. von P. aus den Aminosäuren statt . | | Biosynth.: Meist durch enzymat. „Resektion“ aus Proteinen, die nach Maßgabe des genetischen Codes (Näheres s. dort) u. der Sequenzinformation der Messenger-Ribonucleinsäuren in den Ribosomen gebildet werden, vgl. Peptidhormone. In manchen Fällen findet jedoch durch nicht-ribosomale Enzyme eine Biosynth. von P. aus den Aminosäuren statt . |
| | | | |
| − | Verw.: Zur Hormonsubstitution (Insulin) bzw. als rezeptorselektive Medikamente, die man auch durch gezieltes drug design zu entwickeln hofft. Außerdem werden zur Identifizierung möglicher therapeut., antigener od. anderweitig biolog. aktiver P. mit Hilfe der kombinator. Chemie u. Gentechnologie Zufallsmischungen von P. hergestellt u. in verschiedener Form (z. B. lösl., Polymer-gebunden, auf Phagen) als P.-Bibliotheken verwendet . Der Anw. als Impfstoffe steht noch die mangelnde Reaktion des Immunsystems v. a. auf monomere P. im Wege; zur erfolgreichen Immunisierung müßten die betreffenden P. als B- u. T-Zell-Epitope geeignet sein (vgl. oben). Wegen ihrer Verdaulichkeit u. schlechten Resorption müssen P.-haltige Arzneimittel im allg. parenteral verabreicht, z. B. injiziert od. inhaliert , od. oral als Prodrugs od. Peptidomimetika (peptidähnliche Substanzen) appliziert werden. Zur parenteralen klin. Ernährung kommen synthet. Dipeptide in Betracht .
| |
| | | | |
| | | | |
| | | | |
| − |
| |
| − | Gliederung
| |
| − |
| |
| − | #Proteine
| |
| − | #Aminosäuren
| |
| − | #Peptide
| |
| − | #Entstehung von Peptidbindungen
| |
| | Eine Peptidbindung (-NH-CO-) ist eine Bindung zwischen der Carboxylgruppe einer und der Aminogruppe einer zweiten Aminosäure. | | Eine Peptidbindung (-NH-CO-) ist eine Bindung zwischen der Carboxylgruppe einer und der Aminogruppe einer zweiten Aminosäure. |
| | | | |
Version vom 24. September 2006, 14:40 Uhr

- Johannes
- Mille
Gliederung
- Proteine
- Aminosäuren
- Peptide
- Entstehung von Peptidbindungen
Proteine
(Eiweiße, Eiweißstoffe, Eiweißkörper). Auf Berzelius zurückgehende u. seit Mulder (1838) gebräuchliche u. von griech.: proteuein = „der Erste sein“ abgeleitete Sammelbez. für natürlich vorkommende Copolymere, die sich in der Regel aus 20 verschiedenen a-Aminosäuren (im folgenden: AS) als Monomeren zusammensetzen. Von den nahe verwandten Polypeptiden werden sie aufgrund ihrer mol. Größe unterschieden, wenn auch nicht immer streng abgegrenzt: Ab etwa 100 Monomer-Einheiten (AS-Resten) spricht man meist von Proteinen. Es ergeben sich MR von 10 000 bis mehrere Millionen.
Die Aufeinanderfolge der einzelnen Bausteine (AS-Sequenz, Primärstruktur) unterliegt im allg. keinen offensichtlichen Gesetzmäßigkeiten, so daß potentiell jede Kombination möglich ist. Gäbe es von jedem möglichen Protein-Mol. nur ein Exemplar u. würden nur Mol.-Größen entsprechend 150 AS-Einheiten betrachtet, so ergäbe sich bei 20 verschiedenen AS die unvorstellbar große Zahl von 20150 (eine Zahl mit 195 Stellen) unterschiedlicher Mol., die unser Weltall etwa 1090-mal auffüllen könnten. Die Auswahl aus dieser Fülle treffen die Lebewesen nach Maßgabe der genet. Information (s. bei Biosynth.). Man schätzt, daß in unserem Lebensraum ca. 1011 verschiedene P. vorkommen; ein Höherer Organismus soll ca. 105–106 verschiedene P. enthalten.
Man teilt die P. nach Gestalt u. Verhalten gegen Wasser u. Salze ein in: globuläre od. Sphäroproteine, z. B. Albumine, Globuline, Gluteline, Histone, Prolamine, Protamine, sowie: Skleroproteine od. fibrilläre, Gerüst- od. Faser-Proteine (Gerüst-Eiweiß), z. B. Keratine, Fibroin, Elastin, Collagen. Nach der Zusammensetzung trifft man die Einteilung in einfache P., deren Hydrolyse nur AS gibt, u. zusammengesetzte P. (konjugierte Proteine, veraltet: Proteide), die außer AS für die spezif. Eigenschaften essentielle Nichtproteinkomponenten – die prosthetischen Gruppen (in Klammern; ggf. mit Beisp.) – enthalten: Nucleoproteine (Nucleinsäuren; Chromatin), Glykoproteine (Kohlenhydrate; Lectine, Immunglobuline, Blutgruppensubstanzen), Lipoproteine (Lipide), Phosphoproteine (Phosphorsäure; Casein, Vitelline), Chromoproteine (Farbstoffe; Hämoglobin, Cytochrome, Katalase, Rhodopsin), Metallproteine (Metalle; Caeruloplasmin, Transferrin, Ferredoxin u. a. Eisenproteine) u. a. mehr.
Vork. u. biolog. Bedeutung: P. sind in der belebten Welt allgegenwärtig. Neben Kohlenhydraten u. Fetten (s. Fette u. Öle) sind sie die dritte große Gruppe von Nahrungs- u. Reservestoffen. Auf der Anwesenheit bestimmter P. beruhen Struktur, Funktion u. Stoffwechsel aller lebenden Zellen u. Gewebe; in gewissem Sinn sind die P. die Träger der Lebensfunktionen schlechthin. Man findet sie gleichermaßen in Tieren, Pflanzen u. Mikroorganismen, so z. B. in den Muskeln (Actin, Myoglobin, Myosin), im Blut (Hämoglobin), in Bindegewebe, Sehnen u. Bändern (Collagen, Elastin), im Serum (Fibrinogen, Immunglobuline, s. a. Plasma- u. Serumproteine), in Wolle, Haaren, Hörnern, Hufen, Klauen, Nägeln usw. (Keratine), in den Seidenfäden (Fibroin), in Weichtierschalen (Conchagene), in Knochen (Ossein), in der Milch (Albumine, Casein) usw. – eine vollständige Aufzählung erscheint weder möglich noch sinnvoll. Der P.-Gehalt tier. u. pflanzlicher Organe ist sehr verschieden, z. B.: Fleisch (Muskelgewebe, Rind) 19%, Fisch 16–18%, Knochen (Rind) 30%, Haut 90–97%, Horn, Klauen, Haare 90–100%, Blut (Mensch) 21%, Milch (Mensch) 1%, (Kuh) 3,2%, (Schaf) 5,6%, Eiklar (Huhn) 12–13%. Pflanzliches P. ist vorwiegend in Samen, Knollen usw. gespeichert, z. B. in Getreidekörnern (10–12%), Lupinensamen (37%), Sojabohnen (36%), Kartoffelknollen (nur 2%).
Vielfältig sind auch die Funktionen der P. im Organismus: Als Enzyme (Beisp. s. dort), Transport- u. Speichermol. (Ferritin, Hämoglobin), mol. Motoren (Dynein, Kinesin, Myosin), Gerüstsubstanzen (Skleroproteine, Gerüst-Eiweiß) mit mechan. stützenden Funktionen (Keratine, Collagene, Ossein), in der Immunabwehr (Immunglobuline, Komplement), Hormone (Follitropin, Thyreotropin), Hormon- u. Neurotransmitter-Rezeptoren, Regulatoren (Enzym-Inhibitoren, Transkriptionsfaktoren), Schlangengifte, Bakterientoxine, als Reservestoffe (Gliadin, Zein, Edestin) in Pflanzenorganen usw.
Eigenschaften: Die meist gut wasserlösl. P. (Ausnahmen: Membran-P., s. Membranen, u. Skleroproteine) sind gegen physikal. u. chem. Einwirkung im allg. ziemlich empfindlich. So gerinnt z. B. das Hühner-Eiweiß (Eiklar) oberhalb 65 °C; man bezeichnet diesen Vorgang als Denaturierung. Er beruht auf einer Zerstörung der Raumstruktur der P. unter Aufbrechen eines Teils der schwachen innermol. Wechselwirkungen (vgl. den Abschnitt zur Struktur). Im Gegensatz dazu sind die nativen P. (die man z. B. durch Wasser od. Puffersalz-Lsg. aus den Geweben herauslöst) vermutlich noch in dem gleichen Zustand vorhanden wie im Gewebe selber. Denaturierende Agenzien sind z. B. Guanidiniumchlorid, Harnstoff, Natriumdodecylsulfat, elektr. Ladungen, Säuren (Milchgerinnung infolge Milchsäure-Bildung), Schwermetallsalze usw. Schonendere Ausflockungen ohne bedeutende Denaturierung können z. T. durch Alkohol, Ammoniumsalze u. dgl. erreicht werden. Bei dieser Ausfällung erfolgt eine Schwächung der Hydrathülle der Proteine. Bei der Quellung werden Wasser-Mol. von den P.-Mol. gebunden. Bei vielen globulären P. ist auch eine Kältedenaturierung bekannt, d. h. eine Inaktivierung bei Abkühlung der P.-Lösung.
Aufgrund der ionisierbaren Seitenketten der sauren AS Asparaginsäure u. Glutaminsäure (können Anionen bilden), der bas. AS Lysin, Arginin u. Histidin (können Kationen bilden), sowie der freien Amino- u. Carboxy-Gruppe an den Enden der Polypeptid-Kette kommt dem Protein ein amphoterer Charakter zu, u. es nimmt in Abhängigkeit vom pH-Wert eine jeweils verschiedene elektr. Gesamtladung an; der pH-Wert, bei dem diese verschwindet, heißt isoelektr. Punkt. Bei ihm ist die Wasserlöslichkeit des P. am geringsten.
Struktur: Die Elementaranalyse weist bei P. (neben Sauerstoff, in % Trockengew.) Kohlenstoff (50–52%), Wasserstoff (6,8–7,7%), Stickstoff (15–18%) u. Schwefel (0,5–2%) nach. Häufig findet man auch noch Phosphor, gelegentlich auch Spuren von Eisen, Kupfer, Zink, Mangan, Chlor, Brom, Iod u. dgl., die Begleitsubstanzen (Cofaktoren) angehören.
AS-Zusammensetzung: Der für P. bes. kennzeichnende Stickstoff-Gehalt ist auf ihre Grundbausteine, die AS, zurückzuführen. Mit Hilfe von Säuren, Laugen od. Enzymen (s. Proteasen) lassen sich alle P. nahezu restlos hydrolyt. in AS zerlegen. Die Analyse dieser Hydrolysate ergibt, daß P. – neben selteneren Aminosäuren (s. dort) – immer wieder dieselben 20 AS enthalten, wenn auch in unterschiedlichen Anteilen u. nicht immer alle zugleich, nämlich Glycin (Gly), L-Alanin (Ala), L-Serin (Ser), L-Cystein (Cys), L-Phenylalanin (Phe), L-Tyrosin (Tyr), L-Tryptophan (Trp), L-Threonin (Thr), L-Methionin (Met), L-Valin (Val), L-Prolin (Pro), L-Leucin (Leu), L-Isoleucin (Ile), L-Lysin (Lys), L-Arginin (Arg), L-Histidin (His), L-Asparaginsäure (Asp), L-Asparagin (Asn), L-Glutaminsäure (Glu) u. L-Glutamin (Gln). Alle opt. aktiven AS der P. haben also L-Konfiguration, was im folgenden bei Nennung einzelner Aminosäuren nicht mehr speziell angegeben wird.
Peptid-Bindung: Der Zusammenschluß dieser AS zu den hochmol. P. geschieht durch die Bildung von Säureamid-Bindungen zwischen den Carboxy- u. Amino-Gruppen verschiedener AS-Moleküle. Die Zusammensetzung aus AS u. die Art der Bindung, die man als Peptid-Bindung bezeichnet, haben die P. mit den weniger hochmol. Peptiden gemeinsam. Insbes. unterscheidet man diese nach Anzahl der verknüpften AS-Einheiten als Di-, Tri-, Oligo- (bei bis zu 10 AS-Resten) u. Polypeptide (ca. 10–100 AS-Reste). Demnach kann man Peptide u. P. mit der in Abb. 1 dargestellten Strukturformel charakterisieren, die auch zum Ring geschlossen sein kann, s. Cyclopeptide.
Peptide
(von griech.: peptos = verdaulich). Bez. für durch Peptid-Bindungen Säureamid-artig verknüpfte Kondensationsprodukte von Aminosäuren.
Abb.: Allg. Strukturformel der Peptide.
Bauen sich die Mol. aus 2 Aminosäure-Resten auf, so spricht man von Dipeptiden, bei 3 u. mehr von Tri-, Tetra-, Pentapeptiden etc.; P. mit 2–10 Aminosäure-Resten faßt man als Oligopeptide, solche mit 10–100 als Polypeptide zusammen, doch ist der Übergang von den letzteren zu den höhermol. Proteinen (Eiweißstoffen) nicht genau definiert. P. mit Bindungen zwischen den seitenständigen Amino-Gruppen von Diaminocarbonsäuren (z. B. Lys) u. seitenständigen Carboxy-Gruppen von Aminodicarbonsäuren (z. B. Glu, Asp) statt der üblichen Peptid-Bindungen zwischen a-NH2 u. -COOH nennt man Isopeptide; die von mehrfunktionellen Aminosäuren wie Glu, Asp, Lys, Arg u. Desmosin ausgehenden zusätzlichen Bindungen sind für die Entstehung von P.-Netzstrukturen verantwortlich. P., deren Aminosäure-Sequenz relativ zu einem bestimmten anderen P. die gegenläufige Reihenfolge an Aminosäuren aufweisen, werden als Retropeptide bezeichnet. Zur Schreibung von P.-Formeln benutzt man meist 1- od. 3-Buchstaben-Notationen für die Aminosäuren, s. die Liste dort. Z. B. stehen AG od. Ala-Gly für L-Alanylglycin [H2N–CH(CH3)–CO–NH–CH2–COOH] u. GA od. Gly-Ala für isomeres Glycyl-L-alanin [H2N–CH2–CO–NH–CH(CH3)–COOH]; falls nicht anders gekennzeichnet (etwa durch: Gly¬Ala), steht links die (freie od. protonierte) Amino-Gruppe u. rechts die (freie od. deprotonierte) Carboxy-Gruppe.
Biolog. Bedeutung: Auf die Bedeutung der makromol. P. für pflanzliche u. tier. Organismen wird bei Proteine ausführlich eingegangen. Eine gleichermaßen spezif. Rolle spielen Oligo- u. Polypeptide im tier. Organismus z. B. als Hormone (Peptidhormone), Wachstumsfaktoren, Cytokine, Neurotransmitter u. Neuromodulatoren (Neuropeptide). Für die physiolog. Wirkung der P. ist neben der Konfiguration die Konformation u. die mol. Dynamik von Bedeutung, u. natürlich benötigen die P., um als Mediatoren wirksam werden zu können, spezif. Rezeptoren. Bei der Zell-vermittelten Immunantwort werden Antigene (Fremd-Proteine) von Antigen-präsentierenden Zellen zu P. (Antigen-Peptide, T-Zell-Epitope) abgebaut, von Histokompatibilitäts-Antigenen komplexiert u. so an der Zelloberfläche den T-Lymphocyten zum „Abtasten“ dargeboten; von außen verabreichte P. (peptide feeding) werden ebenfalls präsentiert. Auch von körpereigenen Proteinen abgeleitete Selbst-P. werden präsentiert, was in der Frühphase der T-Zell-Entwicklung für die Entstehung von Selbst-Toleranz von Bedeutung ist. P.-Ester können für süßen (Aspartame®) od. bitteren Geschmack verantwortlich sein, u. wieder andere P. treten als Toxine pflanzlichen od. tier. Ursprungs in Erscheinung. Auch unter den Antibiotika finden sich eine Reihe von P. (Peptid-Antibiotika ), die z. T. Aminosäuren der „unnatürlichen“ D-Konfiguration enthalten, ggf. auch Hydroxycarbonsäuren, die über Esterbindungen verknüpft sind (Peptolide). Viele der physiolog. aktiven P. liegen als homodete od. heterodete Cyclopeptide vor.
Herst.: Auch bei der Synth. ist der zeitliche Aufwand aufgrund der Entwicklung automat. Verf. u. der Festphasen-Technik (Merrifield-Technik) ungleich geringer geworden. Für die Herst. biolog. aktiver u. pharmakolog. nutzbarer P. werden heute neben der chem. Peptid-Synthese in zunehmendem Maße Meth. der Biotechnologie u. Gentechnologie eingesetzt, was z. B. auf dem Gebiet der Peptidhormone bereits zu Erfolgen geführt hat.
Biosynth.: Meist durch enzymat. „Resektion“ aus Proteinen, die nach Maßgabe des genetischen Codes (Näheres s. dort) u. der Sequenzinformation der Messenger-Ribonucleinsäuren in den Ribosomen gebildet werden, vgl. Peptidhormone. In manchen Fällen findet jedoch durch nicht-ribosomale Enzyme eine Biosynth. von P. aus den Aminosäuren statt .
Eine Peptidbindung (-NH-CO-) ist eine Bindung zwischen der Carboxylgruppe einer und der Aminogruppe einer zweiten Aminosäure.
Zwei Aminosäuren können (formal) unter Wasserabspaltung zu einem Dipeptid kondensieren.
Im Beispiel reagieren zwei Moleküle der einfachsten Aminosäure Glycin zu einem Dipeptid: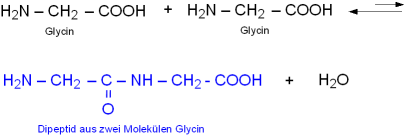
- Struktur der Peptidgruppe
Quelle: Chemie heute Seite 375 Kapitel 19.8,Römpp Lexikon Chemie – Version 2.0, Stuttgart/New York: Georg Thieme Verlag 1999

